
Introduction
Our research goal, broadly stated, is to create new transition metal complexes which have the capability of unusual reactivity, even new reactivity types. These may be Lewis acid/base, hydrogen transfer, or redox. The reactivity always involves changes at the metal, but we have published how our ligand, monoanionic “PNP”, an amide with two phosphine donor “arms,” can also be a reactive functionality: bond-making and -breaking involving substrate. More recently we have moved a bit away from phosphine ligands since these have shown some “fragility” under reactive conditions; nitrogen donors are a solution to this problem. This concept has now been expanded via ligands which are either proton-responsive, changing their character on deprotonation, or having such low lying π* orbitals that they readily store electrons (termed redox active ligands, RAL) for later delivery to coordinated substrate.
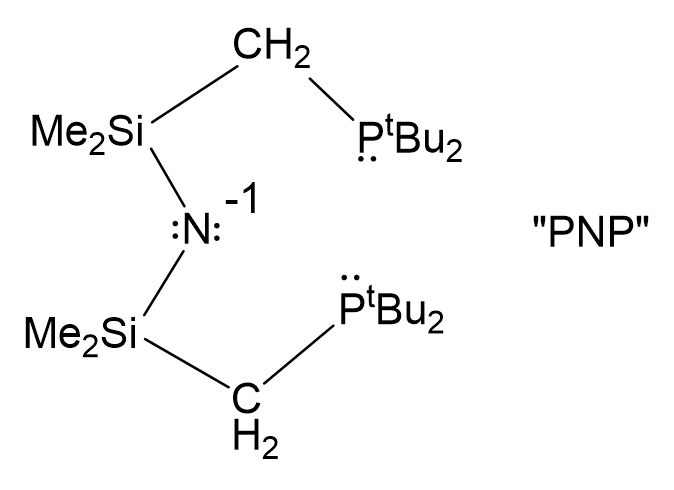
Our metal/substrate reactions are usually the stoichiometric modification of generally unreactive (“inert”) small molecules (N2, N2O, NO, CO2, olefins, but even alkanes and arenes); these are sometimes catalytic. What we strive for are previously unachieved metal complexes and transformations: 14-valence electron complexes, radical hydrides, unusual metal oxidation states, unprecedented ligand binding geometries, very weak substrates, very low coordination numbers, unfavorable metal coordination geometries (e.g. planar d10), multifunctionality, etc. Creating the “unusual” can frequently enable accomplishing the “impossible.” Since we work in these underdeveloped areas, precedent is little guide for us and we often do molecular detective work, using an open mind and variety of physical and spectroscopic techniques: vibrational, X-ray photoelectron and multinuclear NMR spectroscopies, mass spectrometry, EPR, stable isotope labelling and mechanistic tracking. Trying to locate “where the electrons are” in complexes of RAL is greatly facilitated by the technique of X-ray photoelectron spectroscopy, XPS, which has the potential to determine oxidation states of the metal and the nitrogen donors in our unusual complexes. This technique, complemented by scanning tunneling microscopy, STM, of molecules deposited on inert surfaces, has the goal of extending RAL to the field of heterogeneous catalysis. Most often, because we claim to be doing unprecedented things, final proof rests on X-ray diffraction determination of molecular structure: which atoms are bonded together, and whether each bond is single, double, triple, ….. or some weaker noncovalent interaction (e.g. agostic, hydrogen-bonded, or charge-controlled)? Answering these last questions is greatly facilitated by the results of Density Functional Theory (DFT) calculations on structure, bonding, and energy, as well as characterizing transition states of reaction mechanisms. In particularly exciting cases, DFT results precede, and even guide experiment in the most productive directions. “Computational experiments” can predict which substituent will enhance performance.