Biography
Atanu Kumar Das was born in 1982 in West Bengal, India. He completed his Bachelor of Science (BSc) major in chemistry and minor in physics and mathematics, at Ramakrishna Mission Residential College, Narendrapur affiliated under Calcutta University in 2003. In 2005, he received his Master of Science (MSc) in chemistry (specialization in Inorganic chemistry) working on “Synthesis of 21-thiacalix(4)phyrin with two meso sp3 carbons” from Indian Institute of Technology, Bombay, India. He completed his Ph.D. in 2009 under the supervision of Professor Wolfgang Kaim, Institute for Inorganic Chemistry, University of Stuttgart, Germany. His doctoral thesis focused on “Structural, electron transfer and spectroscopic studies of transition metal complexes with redox-active ligands”. Atanu spent one year (2009–2010) as a postdoctoral fellow with Assistant Professor Hong Soon Hyeok at Nanyang Technological University, Singapore working on “Gold-abnormal carbene catalyzed organic transformation”. Atanu joined Professor Kenneth G. Caulton’s group in July 2010 pursuing his second postdoctoral research.
Research Projects
Redox-active Ligand Mediated Bond Making/Breaking Transformations
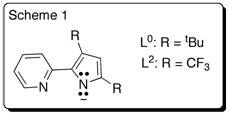
Metal complexes with redox-active ligands have gained much attention not only for their unique and interesting electron transfer behavior but also the prospective role in either stoichiometric or catalytic molecular transformation. Redox-active ligands have more energetically accessible levels that allow redox reactions to change their charge state. Pyridylpyrrolide ligands (scheme 1) have seen considerable recent research from the synthetic perspective, they have not been considered as redox active. We are exploring the 2-pyridylpyrrolide, (Ln)–class of ligands as possible redox active and to expand the redox activity of their metal complexes beyond simple-metal-centered redox chemistry.
Reactivities of [(L0)Ir(Cp*)](BArF4)
A. Activation of oxygen: Electron Buffering from Ligand
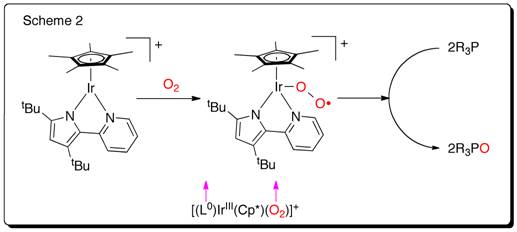
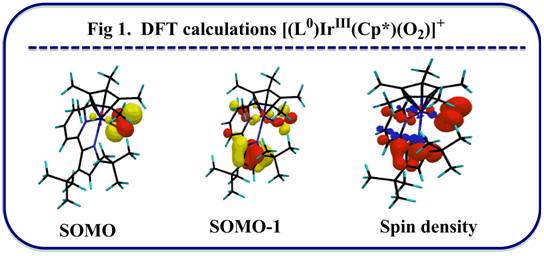
The coordination and activation of dioxygen by transition metal complexes continues to be the focus of considerable interest in attempts to develop new, atom economical oxidation catalysts. However activation by a d6metal [Ir(III) or Ru(II)] complex isn’t well explored so far. A redox non-innocent ligand 2.2’-pyridylpyrrolide that has the potential to store the redox equivalent, both in oxidative and reductive direction, was installed to synthesize a mononuclear [(L0)IrIII(Cp*)Cl] (L0= 2.2’-pyridylpyrrolide) complex to test the reactivity with O2. I find the 18e–complex [(L0)IrIII(Cp*)Cl] is inert to O2, indicating outer sphere electron transfer isn’t viable and O2isn’t strong enough to replace the chloride. However, the unsaturated 16e–species [(L0)IrIII(Cp*)]+obtained from reaction of [(L0)IrIII(Cp*)Cl] with NaBArF4, reacts with O2in the time of mixing (Scheme 2) to form a [(L0)IrIII(Cp*)(O2)]+adduct. The remarkable fact is that the process is irreversible and redox change for the adduct formation occurs at the pyridylpyrrolide. To the best of our knowledge, this is the first example where d6Ir(III)-complex acts as a reducing agent to activate O2. DFT calculations (fig. 1) were conducted by Dr. Kuntal Pal to rationalize the electronic structure of [(L0)IrIII(Cp*)(O2)]+. Different trapping experiments help me to understand the answer to the question, “How many oxygen atom(s) can effectively be transferred to oxidize the substrate?”
Schematic representation of O2activation by pyridylpyrrolide-Ir(III) and oxidation of substrate
B. Heterolytic Cleavage of H2
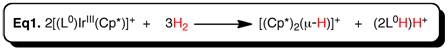
I am interested in extending the redox participation of the pyridylpyrrolide ligand to other redox reagents, a significant one being H2. Questions of interest here are whether H2is a strong enough oxidant to oxidize iridium(III), and what the product of this reaction will be 2H+or dihydride, coordinated dihydrogen, or heterolytic splitting of H2to H–and H+. I find that reaction of dihydrogen (1 atm) with Cp*L0Ir+in CH2Cl2is complete in time of mixing at RT, with rapid color change from green to orange. The iridium containing product is exclusively (Cp*Ir)2(m-H)3+, identified by spectroscopic comparison to an authentic sample. Also produced stoichiometrically (one mole per one Ir) is HL0, the neutral ligand where an H2-derived proton is on the pyrrole nitrogen. Finally, one proton is produced for every two-reagent complexes (eq. 1). This reaction is thus iridium-promotedheterolyticsplitting of three moles of H2. We are using DFT calculation to understand the reaction mechanism.
C. Electrocatalytic CO2Reduction
CO2is the planet’s most important source of carbon and one of the most important atmospheric gases contributing to the greenhouse effect. These are the reasons that its electrochemical reduction into fuels attracts sustained attention. Being motivated by our newly discovered“redox-storage”property of pyridylpyrrolide ligand which serve as“electron-buffer”in small molecule activation e.g., hydrogen oxidation, oxygen activation, we are interested in exploring further the reductive reactivity of M(pyridylpyrrolide)2type of complex.
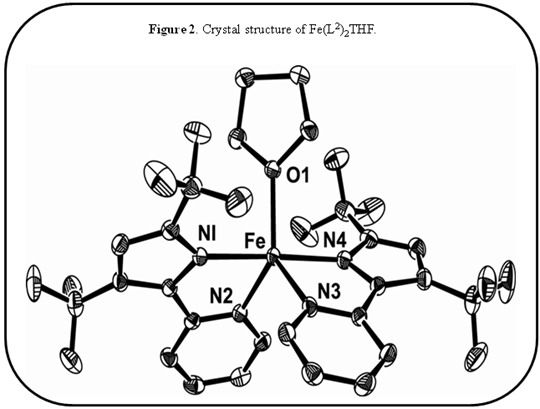
Fe(L2)2(THF) (fig. 2) was synthesized and employed to reduce CO2electrochemically. I have established that Fe(L2)2shows catalytic current flow at -2.42 V (vs. Ag/AgCl) (fig 3) when CV is
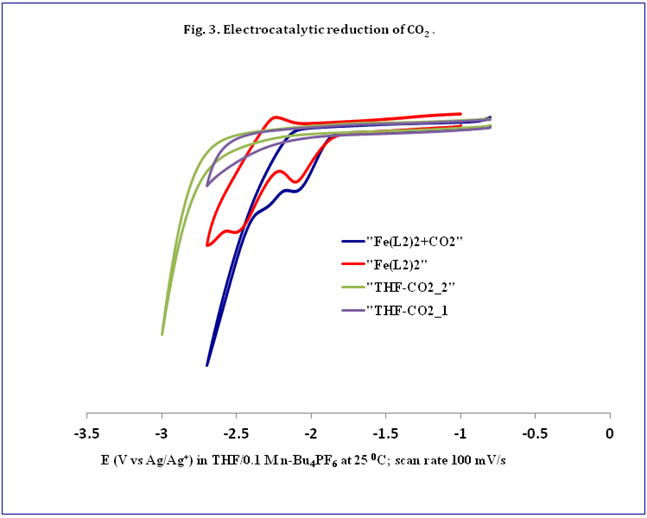
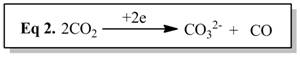
run under 1 atm CO2. Analysis of the colorless precipitate formed concurrently in the electrochemical cell indicates the co-product to be [N(n-bu)4]CO3, hence the product of capture of O2-by CO2in eq 2; [N(n-bu)4]+is the cation in my supporting electrolyte. My current and future plans are to search the liquid phase for other CO2reduction products, particularly more reduced (e.g., alkoxides) and C2species.